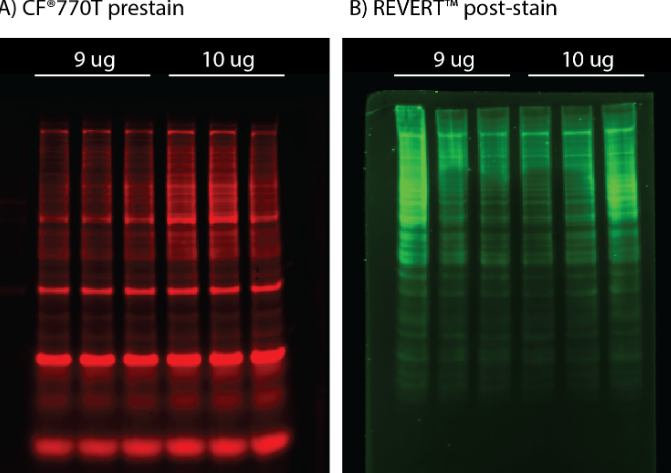
Dal rosa al blu al vicino IR, le macchie di proteine sono disponibili in un’ampia varietà di colori. Come biologi proteici, queste macchie svolgono un ruolo cruciale nel nostro lavoro di laboratorio quotidiano per l’analisi proteica di routine e la normalizzazione della western blot. In effetti, diversi studi hanno scoperto che la normalizzazione per proteina totale è superiore alla normalizzazione con proteine per la pulizia domestica come tubulina o actina 1-3. Tuttavia, ogni colorazione proteica è unica, con vantaggi e svantaggi intrinseci che dovrebbero essere considerati nell’interpretazione dei risultati. In onore della nostra prestaina proteica appena rilasciata, VersaBlot ™, abbiamo deciso di fornire ai nostri lettori una guida per alcune delle più comuni macchie proteiche che include uno sguardo ravvicinato alle loro origini, vantaggi e limiti.
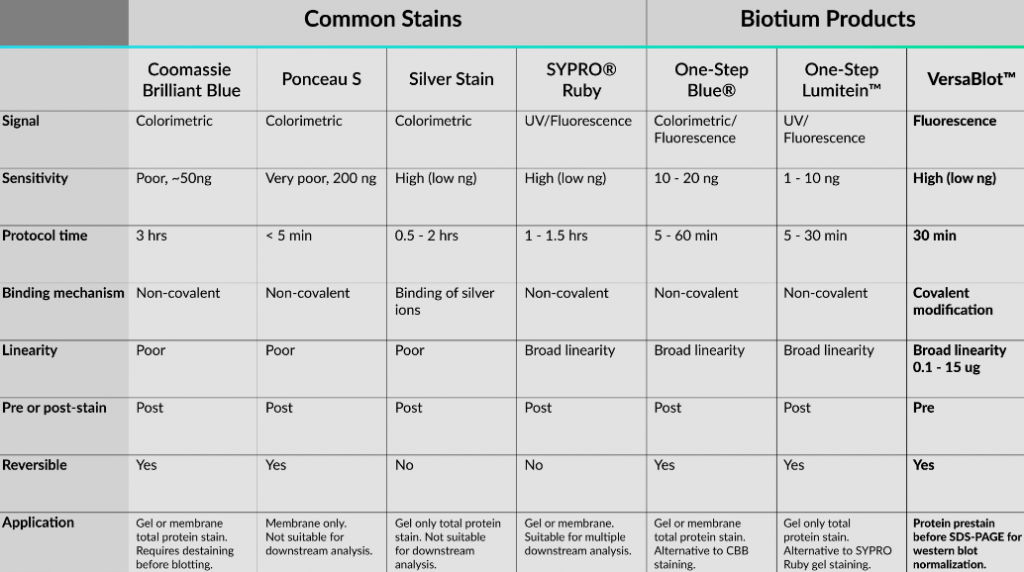
Ecco una tabella utile che riassume le macchie proteiche discusse in questo articolo e le macchie offerte da Biotium.
La “classica” Blue Coomassie
Una guida per la colorazione delle proteine dovrebbe giustamente iniziare con la colorazione proteica più utilizzata, Coomassie Brilliant Blue (CBB). Il CBB fu usato per la prima volta come tintura di lana acida nel 19 ° secolo da un produttore di tinture britannico di nome Levinstein Ltd. Dal 1964, un biochimico australiano di nome Fazekas De St. Growth e colleghi scoprirono la sua applicazione per la colorazione delle proteine dopo l’elettroforesi su gel4. Da allora, sono state sviluppate diverse versioni di CBB, inclusa la formulazione G-250 “colloidale” ampiamente accettata che è più sensibile e ha meno background rispetto alla vecchia forma R-250. Il CBB è molto popolare nei laboratori perché la colorazione è economica e facile da visualizzare. Inoltre, il protocollo di colorazione richiede un solo reagente preparato e si lega alle proteine in modo non covalente attraverso interazioni con residui basici e aromatici. Nonostante questi vantaggi, il CBB manca di sensibilità rispetto ai metodi di colorazione alternativi (es. Colorazione d’argento, fluorescenza) e ha un intervallo lineare ristretto. Inoltre, le membrane trattate con CBB devono essere completamente eliminate o il colorante può interferire con l’analisi a valle.
Vantaggi: economico e facile da visualizzare.
Svantaggi: mancanza di sensibilità, piccolo intervallo lineare, il colorante può interferire con il legame degli anticorpi a valle e l’analisi della spettrometria di massa.
Utilizzare per: Gel o membrana quando è necessaria una colorazione proteica totale economica. Se prevedi di usare il western blot, macchia la membrana dopo il trasferimento e prima del blocco.
Alternative: dai un’occhiata a One-Step Blue® come alternativa più conveniente e versatile alla colorazione con CBB.
Ponceau S, Pink Proteins on the Go
Ponceau, francese per “color papavero”, descrive una famiglia di coloranti Azo rossi usati come coloranti alimentari e macchie di istologia. Ciò che conosciamo come Ponceau nei nostri laboratori è la formulazione Ponceau S, una colorazione proteica totale reversibile per le membrane Western Blot. Come il CBB, Ponceau si lega anche alle proteine in modo non covalente attraverso interazioni ioniche e idrofobiche. Tuttavia, a differenza del protocollo di colorazione più lungo di CBB (almeno 45 minuti più destaining), la colorazione di Ponceau può essere eseguita in pochi minuti e può essere rapidamente eliminata con acqua. Queste comodità consentono a Ponceau di essere un metodo rapido per visualizzare le proteine totali sulle macchie occidentali. Tuttavia, quando Ponceau guadagna in comodità perde in sensibilità. Ponceau è una delle tecniche di colorazione meno sensibili con un rilevamento minimo di 200 ng5. Inoltre, la colorazione è considerevolmente più debole del CBB e può sbiadire gli straordinari, rendendo la normalizzazione meno riproducibile tra gli esperimenti.
Vantaggi: economico, rapido e reversibile.
Svantaggi: scarsa sensibilità e riproducibilità. Non raccomandato per la normalizzazione.
Utilizzare per: colorazione a membrana solo come metodo rapido per verificare l’efficienza del trasferimento di proteine per la western blotting.
La sensibile macchia d’argento
La colorazione dell’argento è considerata lo standard di riferimento per i metodi di rilevazione delle proteine sensibili. La tecnica è stata sviluppata per la prima volta per SDS-PAGE da Switzer et. nel 1979, che ha consentito il rilevamento di sub-nanogrammi e una sensibilità 50 volte maggiore rispetto alla colorazione con CBB6. È interessante notare che il meccanismo alla base di questa sensibilità si basa su una chimica simile allo sviluppo del film in fotografia. I gel proteici sono impregnati di ioni argento (Ag +) che si legano a vari gruppi funzionali di aminoacidi. L’argento viene quindi ridotto a metallo argentato, un processo che è auto-catalitico, consentendo il rilevamento ultrasensibile delle bande proteiche. Sfortunatamente, questa sensibilità non è priva di gravi inconvenienti. Il processo di colorazione può essere molto tecnico e ha una gamma lineare stretta, che porta a una scarsa riproducibilità. Inoltre, la colorazione è irreversibile e solo per uso di gel e quindi non adatta per analisi a valle come spettrometria di massa o western blotting.
Vantaggi: economico con eccellente sensibilità.
Svantaggi: dispendioso in termini di tempo, scarsa riproducibilità e linearità, non compatibile con l’analisi a valle.
Utilizzare per: Gel colorante solo quando è necessaria la sensibilità al di sotto dei nanogrammi per la proteina totale.
SYPRO® Ruby, una nuova era
Nonostante le colorazioni colorimetriche popolari ed economiche, gli svantaggi con sensibilità, linearità e compatibilità con l’analisi a valle hanno portato allo sviluppo di metodi di rilevazione delle proteine più avanzati. Inserisci SYPRO® Ruby, un colorante fluorescente a base di rutenio che è stato sviluppato da Molecular Probes nel 20007. La colorazione SYPRO® Ruby ha permesso la sensibilità alla macchia d’argento ma con un intervallo lineare maggiore e compatibilità con l’analisi a valle. Inoltre, la facilità d’uso e l’elevata riproducibilità di SYPRO® Ruby come colorante proteico totale hanno fornito un metodo superiore per la normalizzazione della western blot rispetto alle proteine domestiche tradizionali1. Oltre un decennio più tardi, sono state sviluppate diverse altre macchie di proteine a base di fluorescenza tra cui LI-COR REVERT ™, Bio-Rad Stain-Free ™ e Amersham ™ Quickstain. Uno svantaggio per le macchie fluorescenti è che sono generalmente più costosi dei metodi colorimetrici e richiedono una termocamera. Tuttavia, man mano che la comunità di ricerca si sposta gradualmente verso tecnologie basate sulla fluorescenza, i dispositivi di imaging a fluorescenza stanno diventando molto più comuni e convenienti.
Vantaggi: comodo, sensibile, ampio intervallo lineare e compatibile con analisi a valle multiple.
Svantaggi: più costoso dei metodi colorimetrici e richiede una termocamera.
Utilizzare per: Esistono formulazioni SYPRO ® Ruby specifiche per il rilevamento di proteine di gel o membrana. Può essere utilizzato per diverse applicazioni tra cui la normalizzazione della western blot e la spettrometria di massa.
Alternative: dai un’occhiata a One-Step Lumitein ™ e One-Step Lumitein ™ UV come alternativa economica e conveniente a SYPRO ® Ruby.
VersaBlot ™, conveniente, altamente lineare e reversibile
Il biotio ha favorito l’avanzamento del rilevamento di proteine fluorescenti con VersaBlot ™, una sostanza altamente lineare, sensibile e reversibile. A differenza delle colorazioni proteiche colorimetriche o fluorescenti in cui viene eseguita la colorazione dopo l’esecuzione del gel, VersaBlot ™ viene aggiunto al campione prima dell’elettroforesi e può essere utilizzato per la normalizzazione della macchia occidentale a valle. Il kit di normalizzazione delle proteine totali VersaBlot ™ viene fornito con due coloranti CF® near-IR scelti per l’emissione in Odyssey® 700 o 800 canali.
Vantaggi: comodo, sensibile con un’ampia gamma lineare. Il segnale può essere disattivato per il rilevamento near-IR multicolore a valle.
Svantaggi: più costoso dei metodi colorimetrici, richiede un riproduttore d’immagini a fluorescenza quasi IR.
Utilizzare per: un metodo conveniente per la normalizzazione della western blot e il rilevamento della proteina near-IR multicolore a valle.
Bring all reagents to room temperature earlier than use. If crystals have shaped in buffer answer, heat to room temperature and blend gently till the crystals have utterly dissolved.
Assay Procedure
Bring all reagents and samples to room temperature earlier than use. It is really useful that each one samples and requirements be assayed in duplicate.
1. Prepare all reagents, working requirements, and samples as directed within the earlier sections.
2. Remove unused microplate strips from the plate body, return them to the foil pouchcontaining the desiccant pack, and reseal.
3. Wash every effectively thrice with Wash Buffer (300 μL/effectively) utilizing a squirt bottle, multi-channel pipette, manifold dispenser or autowasher. Complete elimination of liquid at every step is crucial to good efficiency. Remove any remaining Wash Buffer by aspirating or decanting. Invert the plate and blot it towards clear paper towels.
4. Add 100 μL of every serially diluted protein normal or check pattern per effectively together with a zero normal. Ensure reagent addition is uninterrupted and accomplished inside 15 minutes. Cover/seal the plate and incubate for two hours at room temperature.
5. Repeat the aspiration/wash as in Step 3.
6. Add 100 μL of Detection Antibody in working focus to every effectively. Cover/seal the plate and incubate for 1 hour at room temperature.
7. Repeat the aspiration/wash as in Step 3.
8. Add 200 μL of Substrate Solution to every effectively. Incubate for 20 minutes at room temperature. Protect from gentle.
9. Add 50 μL of Stop Solution to every effectively. If colour change doesn’t seem uniform, gently faucet the plate to make sure thorough mixing.
10. Determine the optical density of every effectively inside 20 minutes, utilizing a microplate reader set to 450 nm.
Calculation of Results
If samples generate values larger than the best normal, dilute the samples and repeat the assay. Calculate the imply absorbance for every normal, management and pattern and subtract common zero normal optical density (O.D.)
Construct a normal curve by plotting the imply absorbance for every normal on the y-axis towards the focus on the x-axis and draw a finest match curve by way of the factors on the graph. Most graphing software program might help make the curve and a 4 parameter logistic (4-PL) often present the perfect match, although different equations (e.g. linear, log/log) will also be tried to see which offers essentially the most correct. Extrapolate the goal protein concentrations for unknown samples from the usual curve plotted.
Each antibody-antigen interplay has distinctive traits. A protocol giving good outcomes for one antibody-antigen pair is perhaps discovered to be unsatisfactory for one more antibody-antigen, even throughout the similar pattern. The interplay of antisera with protein epitopes in Western blotting rely on a lot of components, all contributing to the ultimate sign/noise ratio. Knowledge and management of these components permits for modifications and optimization of the process. Polyclonal antibodies (serum or IgY-fractions from egg yolk) often comprise a lot of totally different antibodies, interacting independently with totally different epitopes (linear epitope covers from Three to 15 amino acids) on the goal used for immunization. This quantity is proscribed for peptide-antigens, but when recombinant or native proteins have been used for immunization, the sign obtained needs to be thought to be cumulative (i.e. attributable to a number of several types of antibodies current within the serum/IgY used).
To obtain reproducible outcomes, we suggest the usage of gloves, forceps, clear and detergent-free laboratory supplies, reagents which haven’t expired, and freshly ready buffers with managed pH. Additionally, it is very important hold protocol volumes, dilutions, and incubation instances the identical. In basic, incubation instances must be as quick as attainable whereas antibody dilutions must be as excessive as attainable.
Agrisera has ready a poster that covers western blot hassle taking pictures, discovered right here. If , you can too request a printed copy.
1. Sample
To be sure that the goal protein is within the beginning materials, it’s important to organize and retailer the samples below non-degrading/aggregating situations. If the goal protein is degraded or not current in adequate quantities within the loaded pattern, it may end up in lack of sign detection. Epitope abundance could be enhanced below altered development situations, by selective tissue preparation, or fractionations of advanced mobile extracts (i.e. organelle preparation). Use the extraction methodology which supplies good yields and constant outcomes. Agrisera has just lately validated Precellys® machine from Bertin Technologies, because it fulfills these standards. For work with organisms that possess strong cell partitions, like diatoms, some suggestions could be discovered right here. Useful reference: “What is the full variety of protein molecules per cell quantity? A name to rethink some printed values.” Milo, 2013.
Sample high quality is of essential significance. As proven within the instance under, samples of Arabidopsis thaliana whole leaf preparations from yr 2008 or 2006 present better degradation of PsbB in comparison with the newer 2009 samples. M denotes molecular weight markers.
The secondary antibody used was Agrisera goat anti-rabbit HRP AS09 602, at 1: 50 000 and chemiluminescent detection reagent.
Protein samples older than 1 yr shouldn’t be used if attainable.
Run pattern extraction buffer (and loading buffer) in a single lane of your gel to examine for contribution of your pattern extraction buffer to the background sign.
Alternative preparation strategies must be thought-about if attainable.
Include optimistic and detrimental controls from the start. Comparison of samples ready from materials containing no goal (e.g. genetic 0-mutants) or greater quantities of the goal (e.g. over-expression mutants) in addition to mobile fractionation (e.g. preparation of varied fractions of organelles) will enable to regulate specificity of obtained indicators.
The size-related identification of a protein-antibody interplay often requires gel separation below totally denaturing situations. Complete discount of intra- and intermolecular S-S bridges ensures the accessibility of the epitope for interplay with antibodies detecting linear epitopes. In this case, decreasing brokers should be added to adequate closing concentrations to each pattern and (often) working buffer. If antibodies acknowledge non-linear epitopes they require conformational integrity of the goal, greatest supplied in non-denaturing PAGE programs, or immuno-histochemistry purposes. Increasing protein loadings would possibly elevate epitope abundance, however most frequently additionally promote non-specific cross-reactions, greater background, and impaired separation.
Avoid protein loadings greater that 5-20 µg/lane for traditional mini-gel programs.
Check separation with stained marker sample or by Coomassie/Silver-staining of the gel. Mixing a pre-stained marker with a marker reacting with the secondary antibody (e.g. MagicMark, Invitrogen) will likely be a bonus for size-determination and can function a management for the visualization assay.
Some extremely hydrophobic membrane proteins would possibly require 2-Eight M urea within the gels and pattern buffer to be stored unfolded throughout separation. TCA/Acetone precipitation may even assist to maintain protein unfolded.If DTT is used as a decreasing agent, it must be freshly ready.
If your polyacrylamide gel is just not polymerizing, wash glass plates rigorously and be certain all grease is eliminated.
3. Protein switch
The protein switch to an acceptable membrane (blotting) with protein-binding capability (nitrocellulose, PVDF) is extremely depending on the biochemical properties of the goal protein. Proteins of upper molecular weight (obvious molecular plenty of >100 kDa) require longer blotting instances than smaller proteins. At low subject strengths (<15 V/cm), the mobility of proteins out of the gel will likely be diminished and proteins is not going to be utterly transferred from the gel. If subject power is simply too excessive (>35 V/cm) proteins would possibly cross by means of the membrane with out binding. The switch time must be as quick as attainable to forestall proteins (particularly decrease molecular weight) from passing by means of the membrane. Place the second membrane and examine the sign power after incubation with major antibody evaluate to the primary membrane. A brief article about it, ready by Agrisera could be discovered right here.
In gradient gels the porosity of the gel is matched with the dimensions of the proteins, that may be a bonus for environment friendly switch.
Check effectivity of a switch by post-transfer staining of the gel (e.g. Coomassie or Silver) or the filter paper (Ponceau, reversible).
Drying of the membrane (air dry between clear sheets of filter-paper) can enhance immobilization of the protein and its denaturation.
For some antibodies, nitrocellulose membrane would possibly work higher in comparison with PVDF and for different antibodies it could be the opposite approach round.
It is essential that blotting gear is nicely rinsed with distilled water after every use and is refrained from contaminating detergents. Foam pads must be cleaned and completely washed. When your pads purchase colours (from plant materials) and/or free stress it’s time to change them.
Do not reuse switch buffers.
The presence of SDS (0.01 to 0.02 %) within the switch buffer will enhance the mobility of proteins (particularly massive proteins) out of the gel and present a detrimental cost to the protein, which can assist to take care of it in its soluble state. At the identical time, SDS will scale back protein binding to the membrane (particularly nitrocellulose) as a consequence of decreased hydrophobicity of the protein.
The presence of alcohol within the switch buffer will lower protein mobility out of the gel. It may even scale back pore dimension of the gel, whereas it is going to enhance binding to nitrocellulose because it removes SDS from proteins and will increase their hydrophobicity. Higher molecular weight proteins may not be transferred utterly if methanol is current within the switch buffer. Change membrane to nitrocellulose, omit methanol from switch buffer, whereas including SDS and rising subject power.
The thickness of the gel will have an effect on protein mobility out of a gel. Thicker gels enable greater loading, however decrease molecular weight proteins would possibly switch much less effectively.
Hydrophobic proteins would possibly migrate additional within the gel than they “ought to” based mostly on their molecular weight. This is as a result of hydrophobic proteins bind extra SDS per amino acid, skewing the cost to mass ration (extra detrimental cost, subsequently they migrate quicker).
4. Blocking
To reduce background staining as a consequence of non-specific membrane-binding of the antibody examined, remaining surfaces of the membrane must be saturated (“blocked”) with proteins that are non-reactive with the antibody. Commonly used are low-fat dry milk powder, casein, IgGs (at 5-10 % however not from a species you’ve gotten your antibodies derived from) or BSA, at 2-5 % w/v. The shortest attainable blocking-time must be decided experimentally on your system. Increasing the blocking-times unnecessarily (e.g. in a single day) or incubation at low temperatures would possibly result in aggregation of blocking protein on the membrane and by this compromise later antibody-epitope interactions. If low background is obtained after 30-60 min of blocking, longer blocking is not going to enhance your outcomes. It must be decided experimentally if including or omitting the blocking reagent within the subsequent antibody incubation steps is an enchancment on the outcomes. Normal buffer and detergent concentrations shouldn’t scale back the impact of the blocking step, nevertheless, accessibility of a goal protein for major antibody could be elevated by a brief “block-free” wash. Each antibody-antigen pair is exclusive and subsequently the blocking protocol must be empirically examined.
As some IgY antibodies would possibly acknowledge milk proteins, BSA would possibly give decrease background than milk powder for these antibodies.
Do not use BSA as blocking agent if BSA-coupled peptides have been used for immunization. BSA must be of excessive purity and IgG-free in order to not intervene with the assay.
As milk incorporates biotin, the usage of milk-power for blocking is incompatible with avidin/streptavidin programs.
If serum is used for blocking, care must be taken to make sure the animal has not been uncovered to (or developed antibodies to) the antigen in query. If that is the case, they could bind to the antigen and stop binding of the first antibody. Purified immunoglobulins are greater high quality blocking reagents. Block with serum from the identical animal species as secondary antibody, NOT the first.
As a protein-free protein various you could block for 1 h at RT with 0.2-0.5 % Tween-20 in PBS adopted by incubation with the first antibodies diluted in 0.25 % Tween-20 in PBS for 1 h at RT.
5. Primary antibody
The major antibody is the foremost determinant of the specificity of the target-recognition. The interplay with the first epitope ought to dominate any cross-reactivity with different epitopes. Commonly used dilutions are between 1:500 to 1:20 000, relying upon the reactivity of the antibody and the detection system used. To examine for specificity of the goal recognition you should utilize (1) one other antibody in opposition to your goal which can bind to different epitopes on a goal protein, (2) run management samples free or depleted of target-proteins, or (3) carry out a peptide competitors assay (for anti-peptide antibody).
If out there, a second antibody identified to react with the pattern can be utilized as a management of subsequent assay steps on a parallel filter in the identical experiment. However, if for instance anti-actin or anti-histone antibody is used it doesn’t evaluate on to lack of sign if aiming to detect a protein of low expression as different detection situations apply in such case. Actin and histone are proteins of excessive expression ranges.
Lyophilized major antibodies incubated for 2-Four h at 4ºC after reconstitution prior to make use of might enhance sign power. Antibodies in numerous codecs (e.g. serum, whole immunoglobulin fraction, affinity purified on a goal) can have numerous necessities for correct storage. For extra data, examine right here.
To diminish unspecific cross-reactions you could pre-adsorb the first antibody (in a single day, 4ºC) with tissue extract which lacks your protein of curiosity (you should utilize a transferred membrane the place the band/space containing your goal protein has been lower out). In case of elevated background indicators, incubate your membrane earlier than any protein is transferred in saturated PBS-milk adopted by protein switch.
Antibodies in opposition to service proteins (KLH, BSA or others) would possibly acknowledge epitopes of different proteins current within the extract. Those anti-carrier antibodies could be eliminated by (1) incubating serum/IgY pattern with a service protein in answer (0.1 % (w/v) or noticed on PVDF membrane or (2) passing serum/IgY pattern by means of the column with certain service protein (use stream by means of fraction in additional experiments).
Primary antibodies saved (at 4°C or -20°C) in answer with blocking reagent usually lose their reactivity. Re-using of unknown major antibody is just not advisable, as most of it could bind in a primary incubation.
Lots of small spots seen on a membrane throughout improvement could be as a consequence of fats in a given serum. To enhance such blots, use milk-based blocking reagents in addition to rising Tween focus in your buffers.
6. Secondary antibody
The secondary antibody needs to be reactive in opposition to the first antibody (e.g. use anti-rabbit to detect major antibodies raised in a rabbit), and is often coupled to an enzyme or dye that permits subsequent visualization. Thus, any non-target binding of the secondary antibody will lead to background (if certain to the membrane as a consequence of inadequate blocking) or false-positive recognition of non-target proteins current on the filter (“cross-reactions”). Secondary antibodies are often used at dilutions of 1:20 000-1:500 000 relying on the sensitivity of the visualization methodology (e.g. enhanced chemiluminescense, ECL or alkaline phosphatase, AP). The optimum dilution of the secondary antibody should be decided experimentally for the detection system used. Different secondary antibodies might even lead to totally different recognition patterns when utilized to the identical pattern. Fro Western, secondary antibodies which can be utilized in excessive dilutions, e.g. >1: 25 000, are advisable.
To examine the contribution of the secondary antibody on the outcomes, you could lower an acceptable sq. of your filter and run it as a parallel management the place the first antibody is omitted (see instance for hassle taking pictures).
If an Ig-reactive marker has been used, the indicators obtained from the marker can function a management of the operate of the secondary antibody, in addition to for dimension dedication.
The sensitivity of the secondary antibodies can differ between numerous producers.
10 μg of mitochondrial fraction from Arabidopsis thaliana(1,3) and Arabidopsis thaliana leaf extract (2,4) have been separated on 10 % gel and blotted on nitrocellulose membrane utilizing moist switch (0.22 % CAPS, pH 11). Filters the place blocked (1.5 h) in 5 % milk in TBST (1X TBS, 0.1% Tween 20), incubated with 1: 1000 anti-COXII antibodies (2 h in TBST) adopted by incubation with 1: 10 000 secondary anti-rabbit (1 h) HRP-coupled antibodies from Agrisera (left panel) and different manufacture (proper panel) and visualized with customary ECL on Kodak autoradiography movie for five s. Antibody in left panel detects goal protein additionally in whole cell extract (2) and can be utilized in greater dilution than utilized 1: 10 000. Agrisera goat anti-rabbit HRP conjugated antibody (AS09 602) can be utilized at following dilutions: 1: 50 000 -1: 90 000 (ELISA), 1 : 75 000 with enhanced ECL and 1: 25 000 with common ECL (WB), 1: 500 -1: 5000 (IHC).
7. Washes
After steps 4, 5 and 6, extra quantities of blocking or antibody must be diminished by washing the membrane in buffer, often the identical buffer used within the creation of blocking or incubating options. Typically, the buffer will embrace a detergent, corresponding to 0.05 % – 0.5 % of Tween20. A Blocking protein in a 1:10 dilution could be added to the wash buffer to assist reduce background. In some protocols, the a number of wash steps omit the detergent within the final step. For causes of reproducibility it is strongly recommended to maintain volumes and instances fixed. The depth of the washing steps could be elevated by (a) elevated instances and volumes, (b) extra modifications of buffer quantity, (c) greater detergent concentrations, and (d) use of stronger detergents (e.g. SDS as an alternative of Tween-20, use high-purity detergents as they may comprise excessive quantities of peroxides and contribute to elevated background). It is sweet apply to confirm that shares of buffers (together with wash buffer) don’t comprise microbial development (seen as cloudiness in answer), as this may contribute to elevated background noise.
8. Detection
Enzymatic detection programs are the preferred ones, and make use of utilization of secondary antibodies conjugated with Alkaline phosphatase (AP or ALP) or horseradish peroxidase (HRP). However, an alkaline phosphatase detection system is a minimum of 10x much less delicate as evaluate to chemiluminescence based mostly detection (ECL). Therefore, utilizing ALP is just not advisable when analysing proteins of low tissue expression. In such circumstances, extra delicate detection programs must be employed, like chemiluminescence. Agrisera affords a set of ECL reagents with two totally different sensitivities: AgriseraECLBright, with mid picogram detection vary and AgriseraECLSuperBright with excessive low femtogram vary. Some laboratories use fluorophore-conjugated antibodies, corresponding to DyLight®, and in such circumstances there is no such thing as a substrate improvement step within the assay. The protocol is shorter, and as a consequence of advances of digital imaging, a number of fluorophores can be utilized in the identical assay.
9. Membrane storage
After western blotting has been accomplished, the membranes (nitrocellulose or PVDF) could be saved for as much as 6 months at room temperature, between sheets of tissue paper (like Kleenex or KimWipes).